Recovery Failure in POTS, Long COVID, ME/CFS and Related Disorders: A Unified Biological Framework
- Graham Exelby
- 6 days ago
- 52 min read

Dr Graham Exelby 2026
Abstract
Successful recovery following infection, injury and physiological stress requires coordinated resolution of inflammatory signalling, restoration of microvascular function, tissue repair and rebuilding of physiological reserve. Although persistent inflammation, endothelial dysfunction, mitochondrial abnormalities and chronic hypoxic signalling have each been implicated in Long COVID, postural orthostatic tachycardia syndrome (POTS), myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and related disorders, these mechanisms are generally considered independently and do not fully explain why recovery fails in susceptible individuals after the initiating insult has resolved.
This review proposes Failed Physiological Resolution as a unifying systems biology framework linking persistent molecular signalling with progressive impairment of adaptive physiology. We propose that Toll-like receptor 4 (TLR4), the receptor for advanced glycation end products (RAGE), nuclear factor-kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), C-C motif chemokine ligand 2 (CCL2) and hypoxia-inducible signalling form an integrated Persistence Network that maintains inflammatory, vascular and metabolic adaptation beyond its normal physiological role.
Rather than functioning as isolated pathways, these signalling systems operate as a self-reinforcing adaptive network that progressively sustains chronic biological activation.
We further propose that persistent signalling is translated into tissue dysfunction through the endothelial–pericyte unit, the principal adaptive regulator of the microcirculation. Progressive disruption of endothelial–pericyte communication impairs adaptive oxygen extraction, promotes extracellular matrix remodelling and compromises lymphatic, glymphatic and interstitial clearance, progressively embedding recovery failure within tissue architecture. The principal physiological consequence is a progressive decline in physiological reserve, reducing the capacity of cardiovascular, neurovascular, metabolic, autonomic, inflammatory and structural systems to respond appropriately to physical, cognitive and orthostatic stress.
Within this framework, chronic illness is interpreted as a disorder of adaptive physiology rather than isolated organ dysfunction. Symptoms emerge when physiological demand exceeds the remaining adaptive reserve, providing a common biological explanation for fatigue, post-exertional symptom exacerbation, autonomic dysfunction, cognitive impairment and exercise intolerance across apparently diverse chronic disorders.
Although requiring prospective validation, this framework integrates molecular signalling, microvascular regulation and systems physiology into a coherent model of recovery biology. By shifting emphasis from persistent disease processes to failure of physiological resolution, it provides a unifying conceptual foundation for understanding Long COVID, POTS, ME/CFS and related disorders, while identifying restoration of adaptive physiology and physiological reserve as central objectives for future mechanistic research and therapeutic development.
Keywords: Failed physiological resolution; Long COVID; Myalgic encephalomyelitis/chronic fatigue syndrome; ME/CFS; postural orthostatic tachycardia syndrome; POTS; physiological reserve; endothelial cells; pericytes; microvascular dysfunction; oxygen extraction; TLR4; RAGE; NF-κB; STAT3; HIF-2α; systems biology.
1. Introduction
Recovery following infection, injury or physiological stress is one of the defining characteristics of healthy biological systems. Acute inflammatory, vascular and metabolic responses are essential for tissue protection and repair, yet they are normally self-limiting, resolving once the initiating insult has subsided. Successful recovery therefore depends not only upon surviving injury but upon restoring physiological homeostasis, rebuilding adaptive capacity and preserving physiological reserve. Failure of these coordinated processes may result in persistent multisystem dysfunction despite apparent resolution of the original trigger.
Such recovery failure is increasingly recognised following viral infection, trauma, surgery, toxic exposure and other major physiological stressors, suggesting that diverse initiating insults may converge upon common biological mechanisms governing recovery rather than disease initiation alone [4,6,9–11].
Long COVID, postural orthostatic tachycardia syndrome (POTS), myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and several related chronic disorders illustrate this clinical challenge. Although these conditions differ in their initiating events and clinical presentation, they share striking clinical features including exercise intolerance, autonomic dysfunction, cognitive impairment, fatigue, sensory hypersensitivity and delayed recovery following physical or cognitive stress. This substantial overlap suggests that common biological mechanisms may underlie apparently distinct disease entities [1–8].
Considerable progress has been made in identifying individual mechanisms contributing to these disorders, including persistent innate immune activation, endothelial dysfunction, autonomic dysregulation, mitochondrial impairment, neuroinflammation, chronic hypoxic signalling and microvascular abnormalities. However, these mechanisms are commonly investigated in isolation and therefore provide only partial explanations for the progressive loss of adaptive capacity observed clinically. A more fundamental biological question remains insufficiently addressed: why do adaptive recovery programmes fail to terminate once the initiating insult has resolved? [16,19,28,38,47]
In this review, we propose that recovery failure represents a distinct systems-level biological state characterised by persistent activation of an integrated Persistence Network, progressive dysfunction of the endothelial–pericyte unit, and gradual depletion of physiological reserve.
Rather than functioning as independent signalling pathways, Toll-like receptor 4 (TLR4), the receptor for advanced glycation end products (RAGE), nuclear factor-kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), C-C motif chemokine ligand 2 (CCL2) and hypoxia-inducible factor-2α (HIF-2α) are proposed to operate as a coordinated signalling network that sustains inflammatory, vascular and hypoxic adaptation beyond its normal physiological role [19,22,24,29,35,38].
Central to this framework is the hypothesis that the endothelial–pericyte unit serves as the principal biological interface through which persistent signalling is translated into impaired tissue physiology. Progressive endothelial–pericyte dysfunction is proposed to reduce adaptive microvascular regulation and oxygen extraction, promote extracellular matrix remodelling, disrupt lymphatic, glymphatic and interstitial clearance, and progressively erode physiological reserve. As reserve declines, the organism becomes increasingly unable to tolerate physical, cognitive and orthostatic stress, creating a self-reinforcing cycle of incomplete recovery and progressive multisystem dysfunction [47,51–55,70–77].
This perspective shifts attention from the initiating insult towards the biology of recovery itself. Rather than viewing chronic illness as the persistence of isolated inflammatory or metabolic abnormalities, recovery failure is proposed to represent progressive impairment of the integrated adaptive systems responsible for restoring physiological homeostasis. Viral infection, trauma, connective tissue disorders, vascular compression syndromes, toxic exposures and other physiological stressors may activate different upstream pathways, yet each has the potential to converge upon the same downstream mechanisms governing adaptive recovery.
Accordingly, this review develops an integrated systems biology framework describing how persistent signalling, endothelial–pericyte dysfunction and progressive depletion of physiological reserve interact to produce chronic recovery failure. By synthesising current evidence across molecular signalling, neurovascular biology, hypoxic adaptation, extracellular matrix remodelling and recovery physiology, we propose a unifying model capable of integrating Long COVID, POTS, ME/CFS and related disorders within a common biological framework.
The therapeutic implications of this model, including targeted modulation of the Persistence Network and restoration of adaptive capacity, are discussed in the companion paper, Modulating the RAAS–RAGE–TLR4–Pericyte Axis in POTS, Long COVID, ME/CFS and Related Disorders.
2. The Persistence Network: Why Adaptive Signalling Persists
2.1 Acute Adaptive Signalling: Designed for Survival
The innate immune response represents one of the most highly conserved biological defence systems, enabling rapid recognition of infection, tissue injury and cellular stress. Pattern-recognition receptors (PRRs), including the Toll-like receptor (TLR) family, recognise pathogen-associated molecular patterns (PAMPs) derived from microorganisms together with endogenous danger-associated molecular patterns (DAMPs) released from damaged or stressed tissues. Activation of these receptors initiates a coordinated programme of inflammatory, vascular and metabolic adaptation designed to contain injury, eliminate pathogens and promote tissue repair [16–20].
Among these receptors, TLR4 occupies a particularly important position because it responds not only to bacterial lipopolysaccharide but also to numerous endogenous danger signals, including HMGB1, S100 proteins, heat-shock proteins and extracellular matrix fragments generated during tissue injury [17–20]. Activation of TLR4 stimulates downstream signalling through MyD88-dependent and TRIF-dependent pathways, resulting in activation of nuclear factor-kappa B (NF-κB), interferon regulatory factors and multiple inflammatory cytokines that coordinate the early host response [17,18,28–30].
Simultaneously, tissue injury results in activation of the receptor for advanced glycation end products (RAGE) through binding of HMGB1, S100 proteins and other endogenous ligands released during cellular stress. Although originally characterised in diabetes, RAGE is now recognised as a central amplifier of sterile inflammation, integrating inflammatory signalling with oxidative stress, endothelial activation and vascular dysfunction [22–27].
The inflammatory response is closely integrated with metabolic adaptation. As oxygen availability becomes limited or tissue metabolic demand increases, stabilisation of hypoxia-inducible factors (HIFs) coordinates transcriptional programmes that maintain cellular survival by promoting angiogenesis, glycolysis and adaptive metabolic reprogramming. Simultaneously, activation of STAT3 and NF-κB regulates inflammatory gene expression, cellular proliferation and tissue repair, while chemokines such as CCL2 recruit monocytes and macrophages essential for resolution of injury [34–46].
Under physiological conditions these signalling pathways are self-limiting. Once pathogens have been eliminated and tissue repair completed, anti-inflammatory mediators, specialised pro-resolving pathways and homeostatic feedback mechanisms progressively terminate inflammatory signalling, restore endothelial function and re-establish tissue homeostasis [16,17,92–97]. Consequently, acute activation of TLR4, RAGE, NF-κB, STAT3, CCL2 and HIF signalling should be regarded as an essential component of normal adaptive biology rather than pathological processes in themselves.
The central question addressed by the present framework is therefore not why these pathways are activated, but why they fail to switch off. We propose that, under certain biological conditions, these normally protective adaptive responses become chronically interconnected, creating a self-reinforcing signalling network that persists long after the initiating insult has resolved. This persistent signalling state forms the basis of the Persistence Network.
2.2 The Persistence Network: From Independent Pathways to an Integrated Signalling Network
Traditional models of chronic inflammatory disease have generally considered TLR4, RAGE, NF-κB, STAT3, CCL2 and hypoxia-inducible signalling as largely independent pathways contributing to specific aspects of disease. Although this reductionist approach has greatly advanced understanding of individual signalling mechanisms, it does not adequately explain why chronic inflammatory states frequently become self-sustaining or why apparently diverse initiating insults converge upon remarkably similar biological phenotypes. Increasing evidence indicates that these pathways exhibit extensive bidirectional communication, forming an integrated signalling network rather than functioning as isolated molecular cascades [17–19,22–24,28–30,34–39].
Within this integrated network, TLR4 functions primarily as the sentinel receptor, detecting pathogen-associated and danger-associated molecular patterns that signal infection or tissue injury. RAGE acts principally as an amplification receptor, responding to endogenous ligands released from damaged cells and sustaining inflammatory activation after the initiating insult has subsided.
Together these receptors initiate and reinforce activation of NF-κB, which serves as the principal transcriptional regulator of acute inflammatory responses. STAT3 integrates inflammatory and reparative signalling, while CCL2 coordinates recruitment of monocytes and macrophages necessary for tissue remodelling. Hypoxia-inducible factors provide metabolic adaptation to reduced oxygen availability, linking inflammatory activation with changes in cellular bioenergetics and tissue oxygen utilisation [17–24,28–46].
Importantly, these signalling pathways are interconnected through multiple positive feedback loops. NF-κB enhances expression of cytokines that activate STAT3; STAT3 prolongs inflammatory transcriptional programmes while interacting with HIF-dependent signalling; hypoxia promotes further NF-κB activation; RAGE activation amplifies oxidative stress and inflammatory signalling; and CCL2-mediated recruitment of inflammatory cells increases production of additional cytokines, DAMPs and reactive oxygen species. Consequently, activation of any one component of the network may progressively reinforce activation of the others, producing a stable biological state that persists independently of the original initiating event [22–24,28–46].
The Persistence Network therefore differs fundamentally from a simple inflammatory cascade. Rather than a linear sequence of molecular events, it represents a self-organising signalling ecology in which multiple interacting pathways collectively maintain chronic adaptive activation. Such biological organisation provides resilience during acute injury by ensuring robust host defence. However, when feedback regulation fails or adaptive signalling remains chronically engaged, the same architecture becomes capable of sustaining persistent inflammatory, vascular and hypoxic adaptation despite resolution of the initiating insult.
This systems perspective also provides a biological explanation for the convergence of apparently unrelated triggers. Viral infection, bacterial illness, mechanical injury, connective tissue disorders, vascular compression syndromes, metabolic stress, toxic exposures and autoimmune activation all generate varying combinations of DAMPs, inflammatory cytokines, oxidative stress and hypoxia. Although these initiating mechanisms differ substantially, each has the potential to activate the same integrated signalling network. Persistent illness may therefore reflect convergence upon a common adaptive programme rather than persistence of the initiating trigger itself.
Within the present framework, the Persistence Network represents the molecular foundation of recovery failure. Its importance lies not in the pathological role of any single signalling pathway but in the capacity of the network as a whole to maintain chronic biological adaptation. The critical question therefore becomes where this persistent signalling is ultimately expressed within tissues. Increasing evidence suggests that the endothelial–pericyte unit represents the principal biological interface through which the Persistence Network is translated into impaired tissue physiology.
2.3 Self-Reinforcing Signalling: The Biological Basis of Persistence
A defining characteristic of the Persistence Network is its capacity to sustain biological activation through multiple interconnected positive feedback mechanisms. Under physiological conditions, inflammatory signalling is tightly regulated by negative feedback pathways that terminate the response once tissue repair has been completed. In contrast, persistent activation of TLR4, RAGE, NF-κB, STAT3, CCL2 and hypoxia-inducible signalling progressively shifts the balance towards self-sustaining activation, allowing adaptive programmes originally intended for short-term survival to persist long after the initiating insult has resolved [16–24,28–46,92–97].
One of the principal mechanisms underlying persistence is the continued generation of danger-associated molecular patterns (DAMPs). Cellular stress, oxidative injury, extracellular matrix remodelling and impaired tissue clearance result in ongoing release of endogenous ligands such as HMGB1, S100 proteins, mitochondrial DNA and matrix degradation products. These molecules continually stimulate TLR4 and RAGE, maintaining innate immune activation even in the absence of persistent infection [19–27].
Persistent inflammatory signalling also promotes chronic oxidative stress, which further amplifies RAGE activation and enhances NF-κB-dependent transcription. NF-κB subsequently increases expression of pro-inflammatory cytokines, chemokines and adhesion molecules, while IL-6-mediated activation of STAT3 prolongs inflammatory gene expression and promotes cellular adaptation rather than resolution. This reciprocal interaction between NF-κB and STAT3 represents one of the principal mechanisms through which acute inflammation evolves into chronic biological activation [28–37].
Hypoxia further stabilises the Persistence Network. Reduced efficiency of microvascular oxygen extraction increases HIF activation, promoting metabolic adaptation, angiogenic signalling and inflammatory gene expression. In turn, HIF signalling interacts closely with both NF-κB and STAT3, reinforcing inflammatory activation while maintaining cellular survival under conditions of reduced oxygen availability. Hypoxia therefore functions not simply as a consequence of inflammation but as an active participant in sustaining the persistent adaptive state [38–42].
Recruitment of monocytes and macrophages through CCL2 provides an additional amplification mechanism. Activated myeloid cells generate cytokines, reactive oxygen species and additional DAMPs that perpetuate endothelial activation, extracellular matrix remodelling and hypoxic adaptation. Under physiological conditions these cells contribute to tissue repair and resolution. When signalling remains persistent, however, they become important contributors to the chronic inflammatory microenvironment [43–46].
Importantly, the Persistence Network is proposed to function as an adaptive biological system rather than a pathological signalling error. Each component fulfils an essential physiological role during host defence and tissue repair. Persistence develops because normal adaptive programmes fail to terminate appropriately, resulting in prolonged activation of mechanisms originally designed to preserve tissue survival. Recovery failure therefore reflects failure of physiological resolution rather than inappropriate activation of otherwise abnormal pathways.
This perspective helps explain why persistent illness frequently continues despite clearance of the initiating pathogen or resolution of the original injury. Once established, the Persistence Network becomes increasingly capable of maintaining its own biological activity through reciprocal amplification between inflammatory signalling, hypoxic adaptation, oxidative stress and impaired tissue repair. Chronic disease is therefore proposed to represent a state of persistent adaptive signalling maintained by the architecture of the network itself.
2.4 Progressive Hypoxic Adaptation: From Acute Survival to Chronic Biological Reprogramming
Hypoxia is one of the most powerful physiological stimuli regulating tissue adaptation. During acute injury, transient reductions in oxygen availability activate hypoxia-inducible factors (HIFs), initiating transcriptional programmes that preserve cellular survival through increased glycolysis, angiogenesis, erythropoiesis and metabolic adaptation. These responses are essential components of normal recovery, allowing tissues to maintain function while oxygen delivery and microvascular integrity are restored [38–42].
Under physiological conditions, hypoxic signalling is transient. As tissue oxygenation improves and normal microvascular function is re-established, HIF activation declines and cellular metabolism progressively returns towards oxidative phosphorylation. Persistent activation of hypoxia-inducible pathways therefore implies either continuing impairment of oxygen availability or failure of the adaptive systems responsible for restoring tissue oxygen homeostasis [38–42].
Within the Recovery Failure framework, persistent dysfunction of the Persistence Network progressively impairs adaptive oxygen extraction, resulting in relative tissue hypoxia despite preserved macrocirculatory perfusion. Rather than representing complete oxygen deprivation, this state reflects an inability of the microcirculation to increase oxygen extraction sufficiently to meet changing metabolic demand.
Consequently, tissues remain chronically exposed to low-grade hypoxic signalling even when conventional measures of systemic oxygen delivery remain normal.
Increasing evidence suggests that chronic hypoxia is characterised by a gradual shift in the dominant hypoxic response. HIF-1α predominates during acute hypoxic stress, promoting glycolytic metabolism, inflammatory activation and rapid adaptive responses that support short-term survival.
As hypoxia becomes persistent, however, HIF-2α assumes increasing importance, promoting vascular remodelling, endothelial adaptation, extracellular matrix reorganisation and longer-term tissue survival. This transition represents a change in biological strategy—from immediate cellular protection towards sustained adaptation to chronic physiological stress [38–42].
Importantly, HIF-2α does not function independently of the Persistence Network. Extensive crosstalk exists between HIF signalling, NF-κB and STAT3, allowing inflammatory, metabolic and hypoxic pathways to reinforce one another. Chronic activation of STAT3 promotes prolonged HIF-dependent transcription, while persistent hypoxia further amplifies inflammatory signalling and endothelial activation. The resulting interactions progressively stabilise the adaptive state, making spontaneous biological resolution increasingly difficult [34–42].
This progressive hypoxic adaptation has important consequences for tissue physiology. Sustained HIF-dependent signalling alters endothelial behaviour, pericyte function, extracellular matrix turnover and angiogenic responses while increasing reliance upon glycolytic metabolism. Although these changes initially preserve tissue viability, they progressively reduce metabolic efficiency, impair oxygen extraction and diminish physiological flexibility. Adaptive programmes that are beneficial during acute injury therefore become increasingly maladaptive when maintained chronically.
Chronic hypoxia should therefore be regarded not simply as a downstream consequence of persistent disease but as an active participant in recovery failure. Progressive HIF-dependent reprogramming links persistent molecular signalling with the structural and functional changes that develop within the microcirculation. This provides the biological bridge between the Persistence Network described in this section and the endothelial–pericyte dysfunction that forms the focus of Section 3, The Endothelial–Pericyte Unit: Translating Persistent Signalling into Tissue Dysfunction.
2.5 The Persistence Network as a Systems Biology Model
Biological systems rarely function as isolated molecular pathways. Instead, they comprise highly interconnected regulatory networks in which numerous signalling pathways continuously exchange information, adapt to changing environmental conditions and collectively determine physiological behaviour. The response of such systems cannot be predicted simply by examining the activity of individual components because the biological properties of the network emerge from the interactions between its constituent pathways. This concept forms one of the central principles of modern systems biology [87,88].
The Persistence Network proposed in this review should therefore be regarded as an emergent adaptive network rather than a collection of independent inflammatory pathways. TLR4, RAGE, NF-κB, STAT3, CCL2 and HIF signalling each fulfil distinct physiological roles, yet their biological importance lies primarily in their continuous interaction. Through reciprocal amplification and multiple positive feedback loops, these pathways collectively generate a stable adaptive state capable of sustaining inflammatory, vascular and metabolic responses long after the initiating insult has resolved.
This systems perspective also explains why chronic disease frequently demonstrates non-linear behaviour. Relatively small physiological stressors may produce disproportionately large clinical deterioration because the Persistence Network already exists close to a critical adaptive threshold. Minor increases in inflammatory activity, hypoxia or oxidative stress can therefore trigger widespread changes throughout the network, producing substantial reductions in physiological performance without requiring major structural tissue injury.
Conversely, improvement in a single component of the network may not immediately produce clinical recovery if multiple reinforcing pathways continue to maintain the persistent adaptive state. Restoration of adaptive physiology is therefore unlikely to depend upon normalising one molecular pathway alone. Rather, recovery probably requires progressive restoration of the regulatory interactions that normally allow inflammatory signalling, endothelial activation and metabolic adaptation to resolve once tissue repair has been completed.
Importantly, this framework also provides an explanation for the convergence of apparently diverse chronic disorders. Viral infection, bacterial illness, trauma, connective tissue disorders, vascular compression syndromes, toxic exposures and autoimmune activation differ substantially in their initiating mechanisms. However, each may generate combinations of inflammatory activation, DAMP release, oxidative stress and impaired oxygen homeostasis capable of activating the same integrated adaptive network. Chronic illness therefore reflects convergence upon a common biological programme rather than persistence of the initiating trigger.
The Persistence Network should therefore be regarded as the molecular engine of recovery failure. However, persistent signalling alone does not directly produce clinical disease. The critical question becomes where this persistent adaptive programme is translated into altered tissue physiology. Increasing evidence suggests that this occurs principally within the endothelial–pericyte unit, which integrates inflammatory signalling, oxygen extraction and microvascular adaptation at the level of the capillary circulation.
Table 1. Functional Roles Within the Persistence Network
Component | Primary Physiological Role | Contribution to Persistent Signalling |
TLR4 | Detects infection and tissue injury | Initiates innate immune activation through recognition of PAMPs and DAMPs |
RAGE | Amplifies tissue stress responses | Sustains inflammatory signalling following cellular injury |
NF-κB | Coordinates inflammatory gene transcription | Maintains cytokine production and inflammatory amplification |
STAT3 | Integrates inflammatory and reparative responses | Promotes prolonged adaptive signalling and tissue remodelling |
CCL2 | Recruits monocytes and macrophages | Sustains immune-cell recruitment and chronic inflammatory activation |
HIF-1α / HIF-2α | Coordinates adaptation to hypoxia | Maintains metabolic and vascular adaptation during chronic oxygen stress |
Footnote: HIF-1α predominant acutely; HIF-2α increasingly dominant in chronic/persistent states
2.6 Transition paragraph
Collectively, these pathways form the core of the Persistence Network. While each fulfils a distinct biological role, their importance lies in their interaction rather than their individual actions. Together they create a self-reinforcing signalling ecology capable of sustaining inflammatory, vascular, metabolic and hypoxic adaptation long after the initiating insult has resolved. The next question therefore becomes where this persistent signalling is ultimately expressed biologically.
Increasing evidence suggests that the endothelial–pericyte unit represents the principal interface through which the Persistence Network is translated into impaired oxygen extraction, reduced physiological reserve and recovery failure.
3. The Endothelial–Pericyte Unit: Translating Persistent Signalling into Tissue Dysfunction
The Persistence Network described in the preceding section explains how inflammatory, vascular and hypoxic signalling may remain chronically activated following infection, injury or other physiological stress. Persistent molecular signalling alone, however, cannot explain the characteristic clinical manifestations of recovery failure. Chronic illness develops only when these signalling pathways are translated into alterations in tissue physiology that impair adaptive biological function.
We propose that this translation occurs principally within the endothelial–pericyte unit, the fundamental regulatory interface of the microcirculation. Rather than functioning simply as structural components of capillary walls, endothelial cells and pericytes form an integrated adaptive system that continuously regulates tissue oxygen delivery, capillary recruitment, vascular permeability, inflammatory signalling, extracellular matrix remodelling and tissue repair. Their strategic location at the interface between circulating blood and tissue metabolism places them in a unique position to integrate inflammatory, metabolic and hypoxic signals while coordinating local physiological adaptation [47–60].
This perspective represents an important conceptual shift. Endothelial dysfunction has long been recognised in Long COVID, POTS, ME/CFS and numerous chronic inflammatory disorders. Similarly, increasing evidence implicates pericyte dysfunction in neurovascular disease, microvascular instability and impaired blood–brain barrier integrity. These abnormalities have generally been considered independent pathological findings. Within the Recovery Failure framework, however, they are interpreted as coordinated consequences of persistent adaptive signalling rather than isolated pathological processes [47–60].
Persistent activation of TLR4, RAGE, NF-κB, STAT3, CCL2 and HIF-dependent pathways progressively alters endothelial–pericyte communication, impairing the adaptive regulation of capillary function. Initially these changes remain functional, reducing the efficiency with which the microcirculation responds to increasing physiological demand. As signalling persists, extracellular matrix remodelling, altered tissue mechanics and impaired interstitial clearance progressively stabilise this adaptive state, embedding recovery failure within the tissue microenvironment [47–69].
Importantly, the principal consequence of endothelial–pericyte dysfunction is not simply impaired blood flow, but reduced adaptive microvascular regulation. Healthy tissues continuously adjust capillary recruitment and oxygen extraction according to changing metabolic requirements. Persistent dysfunction of the endothelial–pericyte unit progressively limits this adaptive flexibility, reducing the capacity of tissues to increase oxygen extraction during physical, cognitive and orthostatic stress. This distinction provides the physiological bridge between persistent molecular signalling and progressive depletion of physiological reserve.
Accordingly, the endothelial–pericyte unit is proposed to represent the principal biological interface through which the Persistence Network is translated into chronic multisystem dysfunction. The following sections examine how progressive disruption of this adaptive microvascular system impairs oxygen extraction, remodels tissue architecture and ultimately reduces the physiological reserve necessary for successful recovery.
3.1 The Endothelial–Pericyte Unit: Master Regulator of Microvascular Adaptation
The microcirculation is the principal site of oxygen exchange, nutrient delivery and metabolic waste removal. Although often viewed simply as the terminal component of the vascular tree, the capillary circulation is a highly dynamic adaptive system that continuously matches tissue perfusion to changing metabolic demand. This regulation depends upon the close functional integration of endothelial cells, pericytes,
extracellular matrix components and adjacent parenchymal cells, collectively forming the endothelial–pericyte unit. Rather than serving purely structural roles, these cells coordinate local blood flow, vascular permeability, inflammatory signalling and tissue repair, thereby maintaining physiological homeostasis across multiple organ systems [47–60].
Endothelial cells provide the primary interface between circulating blood and the surrounding tissues. In addition to regulating vascular permeability, they synthesise vasoactive mediators including nitric oxide, prostacyclin and endothelin, coordinate leukocyte trafficking and participate actively in inflammatory signalling. Healthy endothelial function is therefore essential not only for maintaining vascular integrity but also for rapidly adapting tissue perfusion to fluctuating physiological demand [56–60].
Embedded within the capillary basement membrane, pericytes provide the complementary regulatory component of the microcirculation. Through extensive physical and biochemical communication with endothelial cells, pericytes regulate capillary stability, angiogenesis, blood–brain barrier integrity, extracellular matrix turnover and local haemodynamic adaptation. Increasing evidence indicates that pericytes also influence capillary recruitment and regional blood flow distribution, enabling oxygen delivery to be closely matched to tissue metabolic requirements [51–55].
Importantly, endothelial cells and pericytes function as an integrated biological unit rather than as independent cell populations. Bidirectional communication occurs through direct cell–cell contact, paracrine signalling, growth factor networks and interactions with the surrounding extracellular matrix. This integrated regulatory system continuously responds to inflammatory mediators, mechanical forces, oxygen tension and metabolic activity, allowing the microcirculation to adapt rapidly to changing physiological conditions [51–60].
Within the Recovery Failure framework, persistent activation of the Persistence Network progressively disrupts this adaptive communication. Chronic inflammatory signalling, oxidative stress and hypoxic adaptation impair endothelial function while simultaneously altering pericyte behaviour, reducing the efficiency with which the microcirculation adjusts to increased physiological demand. Initially these abnormalities remain functional and potentially reversible. As persistent signalling continues, however, alterations in extracellular matrix organisation, capillary architecture and tissue mechanics progressively stabilise the dysfunctional adaptive state [22–46,61–69].
A key feature of this model is that microvascular dysfunction should not be equated simply with reduced perfusion. Resting blood flow may remain relatively preserved while the capacity to recruit additional capillary exchange surface during physiological stress becomes progressively impaired. Consequently, tissues increasingly lose the ability to augment oxygen extraction during exercise, orthostatic challenge, cognitive activity or inflammation, despite apparently normal macrocirculatory haemodynamics. This distinction provides an important physiological explanation for the discrepancy frequently observed between relatively normal resting investigations and marked functional impairment during stress.
The endothelial–pericyte unit should therefore be regarded as the principal adaptive regulator of tissue oxygen extraction. By integrating inflammatory signalling, vascular regulation, extracellular matrix dynamics and metabolic demand, it determines the efficiency with which tissues respond to physiological stress. Persistent dysfunction of this integrated regulatory system provides the mechanistic bridge between molecular signalling and progressive depletion of physiological reserve that characterises recovery failure.
3.2 Pericytes: The Adaptive Cells of the Microcirculation
Although endothelial dysfunction has been recognised in many chronic inflammatory disorders, increasing evidence suggests that pericytes play an equally important role in regulating adaptive tissue physiology. Embedded within the capillary basement membrane and communicating continuously with adjacent endothelial cells, pericytes are uniquely positioned to integrate inflammatory, metabolic and hypoxic signals while coordinating local microvascular adaptation. Their importance extends well beyond structural support, encompassing regulation of capillary blood flow, vascular permeability, extracellular matrix homeostasis, angiogenesis and tissue repair [51–55].
Unlike vascular smooth muscle cells, which regulate the calibre of larger resistance vessels, pericytes operate at the level of the capillary bed where oxygen exchange occurs. Through dynamic interactions with endothelial cells, pericytes influence capillary recruitment, optimise regional perfusion and facilitate efficient oxygen extraction according to local metabolic demand. This adaptive regulation is particularly important during exercise, orthostatic stress, cognitive activity and tissue repair, when rapid adjustment of capillary function is required to maintain physiological homeostasis [47–60].
Pericytes are also highly responsive to the molecular signals comprising the Persistence Network. Cytokines generated through NF-κB and STAT3 activation, DAMPs acting through TLR4 and RAGE, oxidative stress and sustained hypoxia all modify pericyte behaviour. During acute injury these responses are beneficial, promoting vascular stabilisation, angiogenesis and coordinated tissue repair. When signalling remains persistent, however, the same adaptive programmes progressively become maladaptive, reducing the capacity of pericytes to regulate capillary function effectively [22–46,51–55].
One of the earliest consequences of persistent pericyte activation is disruption of communication within the endothelial–pericyte unit. Altered signalling impairs coordinated regulation of vascular tone, endothelial permeability and capillary recruitment, while promoting extracellular matrix remodelling and changes in basement membrane composition. These abnormalities reduce the flexibility of the microcirculation, limiting its ability to adapt to changing physiological demands without necessarily producing overt structural vascular disease [51–69].
Beyond their vascular functions, pericytes participate actively in tissue homeostasis through regulation of inflammatory signalling, extracellular matrix turnover and stem-cell niches. Within the central nervous system they are essential for maintenance of the blood–brain barrier, neurovascular coupling and cerebral metabolic homeostasis. Similar functions are increasingly recognised in the kidney, retina, myocardium, skeletal muscle and pulmonary microcirculation, suggesting that pericyte dysfunction provides a biologically plausible explanation for the multisystem nature of chronic recovery-failure disorders [52–60].
The Recovery Failure framework proposes that pericyte dysfunction is predominantly functional before it becomes structural. Early impairment reflects altered cellular signalling and adaptive regulation rather than irreversible cell loss. As persistent signalling continues, however, chronic hypoxia, extracellular matrix remodelling and sustained inflammatory activation progressively stabilise the dysfunctional phenotype, reducing the capacity for spontaneous biological recovery. This distinction has important therapeutic implications because restoration of adaptive pericyte function may be achievable before extensive structural remodelling becomes established.
Accordingly, pericytes should be regarded as the adaptive cells of the microcirculation. By integrating inflammatory signalling with vascular regulation, oxygen extraction and tissue repair, they determine the efficiency with which tissues respond to physiological stress. Persistent dysfunction of this adaptive cellular network provides the critical biological bridge between molecular persistence and impaired physiological reserve.
Figure 1. The Endothelial–Pericyte Unit: Translating Persistent Signalling into Progressive Physiological Reserve Loss
Persistent signalling is translated into tissue dysfunction through the endothelial–pericyte unit. Chronic activation of TLR4, RAGE, NF-κB, STAT3 and HIF-2α progressively impairs adaptive microvascular regulation, reducing oxygen extraction and initiating a progressive loss of physiological reserve. Within the Recovery Failure framework, the endothelial–pericyte unit represents the principal biological interface linking persistent molecular signalling with multisystem dysfunction.
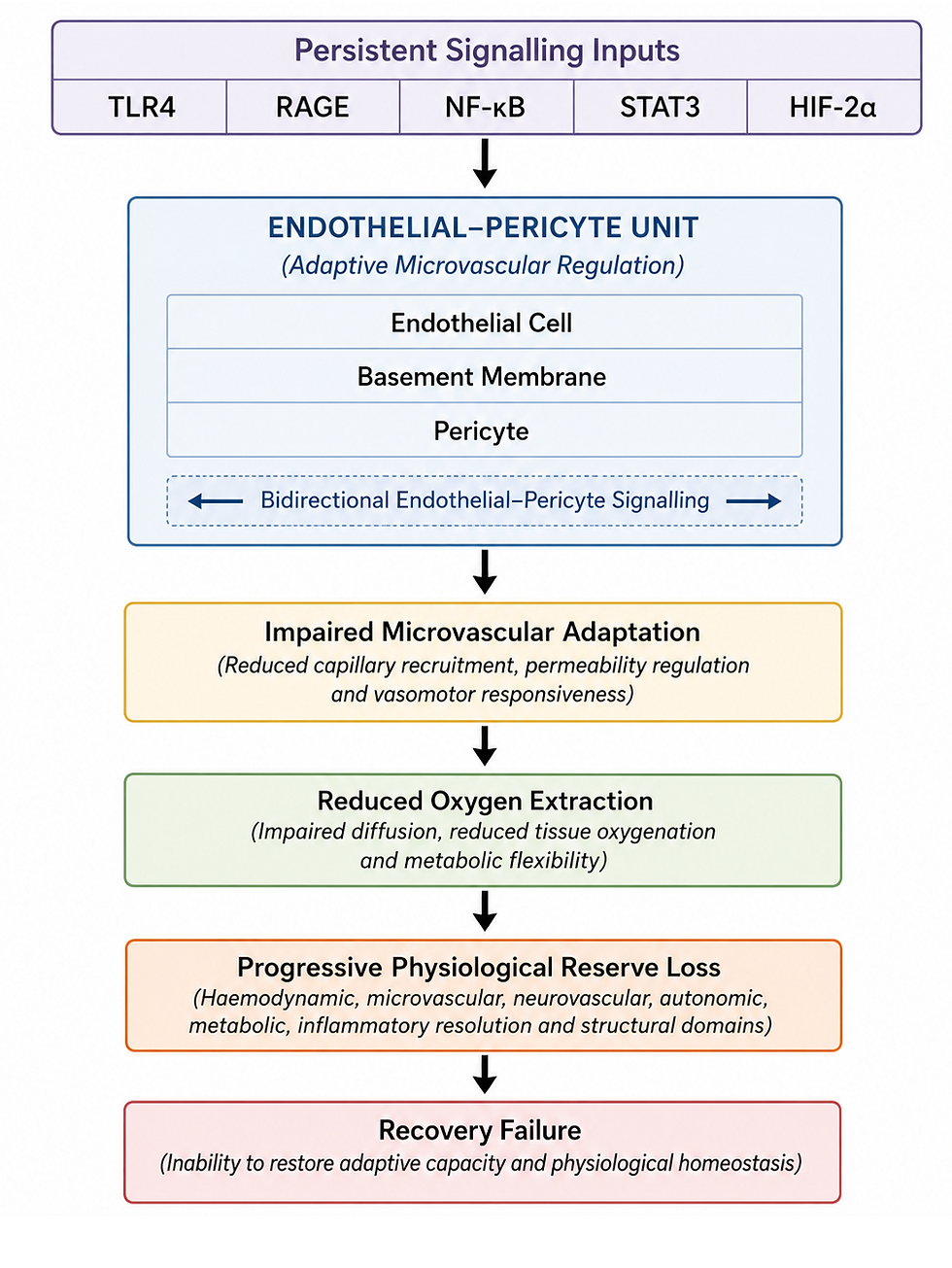
3.3 Impaired Oxygen Extraction: The Functional Consequence of Endothelial–Pericyte Dysfunction
Efficient tissue oxygenation depends upon more than adequate blood flow. Oxygen must be delivered to the microcirculation, distributed appropriately within the capillary network and extracted efficiently according to local metabolic demand. While systemic haemodynamics determine the quantity of oxygen delivered to an organ, the endothelial–pericyte unit determines how effectively that oxygen is transferred to the tissues where it is required. Consequently, tissue oxygenation is fundamentally a function of microvascular adaptability, rather than blood flow alone [47–60,89–91].
Under physiological conditions, increases in metabolic demand trigger rapid adjustments throughout the microcirculation. Capillary recruitment increases, endothelial signalling optimises vascular tone, and pericytes coordinate redistribution of blood flow towards metabolically active regions. These highly integrated responses maximise oxygen extraction while preserving tissue homeostasis during exercise, orthostatic stress, cognitive activity and tissue repair. Once demand falls, adaptive responses rapidly resolve and baseline physiology is restored [47–60,89–91].
Within the Recovery Failure framework, persistent activation of the Persistence Network progressively disrupts these adaptive mechanisms. Endothelial dysfunction reduces nitric oxide bioavailability and impairs vascular responsiveness, while persistent pericyte activation limits the flexibility of capillary recruitment. Simultaneously, extracellular matrix remodelling alters the physical environment surrounding the microcirculation, increasing diffusion distances and reducing the efficiency of oxygen exchange. The combined effect is a progressive reduction in the capacity of tissues to increase oxygen extraction during periods of physiological stress [22–69].
Importantly, impaired oxygen extraction differs fundamentally from impaired oxygen delivery. Large-vessel perfusion may remain normal, arterial oxygen saturation may be preserved, and conventional cardiovascular investigations may demonstrate little abnormality. Nevertheless, tissues may experience functional hypoxia because the adaptive mechanisms responsible for matching oxygen extraction to metabolic demand have become progressively compromised. This distinction provides a physiological explanation for the frequent discrepancy between relatively normal resting investigations and profound functional limitation observed in patients with chronic recovery-failure disorders.
Persistent impairment of oxygen extraction has important biological consequences. Even modest reductions in tissue oxygen availability reinforce hypoxia-inducible signalling, promoting continued activation of HIF-dependent transcription, endothelial activation and extracellular matrix remodelling. Reduced oxygen utilisation also increases metabolic stress, further amplifying inflammatory signalling through NF-κB and STAT3. Consequently, impaired oxygen extraction functions both as a downstream consequence of the Persistence Network and as an important mechanism maintaining its activity [28–42].
This model also explains the characteristic clinical observation that many patients function reasonably well at rest yet deteriorate disproportionately during relatively minor physiological challenges. Basal metabolic requirements may still be met despite impaired adaptive capacity. However, as physical activity, orthostatic stress, cognitive effort or systemic inflammation increase tissue oxygen demand, the diminished capacity to augment oxygen extraction becomes increasingly limiting. Symptoms therefore emerge when physiological demand exceeds adaptive oxygen extraction reserve, rather than simply because resting tissue perfusion is reduced.
Accordingly, impaired oxygen extraction represents the principal physiological consequence of endothelial–pericyte dysfunction. Rather than viewing chronic illness as a disorder of inadequate blood flow, the Recovery Failure framework proposes that the defining abnormality is loss of adaptive oxygen extraction, progressively reducing tissue resilience and initiating depletion of physiological reserve.
3.4 Extracellular Matrix Remodelling: Embedding Recovery Failure into Tissue Architecture
The extracellular matrix (ECM) is increasingly recognised as a dynamic biological regulator rather than simply a structural scaffold. In addition to providing mechanical support, the ECM continuously regulates cell adhesion, mechanotransduction, angiogenesis, inflammatory signalling and tissue repair through reciprocal interactions with endothelial cells, pericytes, immune cells and resident stromal cells.
Normal recovery requires tightly coordinated ECM remodelling that restores tissue architecture once injury has resolved. Persistent activation of the Persistence Network, however, progressively alters these adaptive processes, allowing chronic biological signalling to become incorporated into the tissue microenvironment [61–69].
During acute injury, controlled ECM remodelling facilitates vascular repair, immune-cell migration and restoration of tissue integrity. Matrix synthesis and degradation remain tightly balanced through coordinated regulation of fibroblasts, pericytes, matrix metalloproteinases and growth factor signalling. As inflammatory activity resolves, normal tissue architecture is progressively re-established, restoring both structural integrity and physiological flexibility [61–69,92–97].
Within the Recovery Failure framework, persistent inflammatory, hypoxic and metabolic signalling progressively disrupts this balance. Sustained activation of NF-κB, STAT3 and HIF-dependent pathways promotes ongoing extracellular matrix turnover, altered collagen organisation and changes in basement membrane composition. Pericytes increasingly adopt matrix-remodelling phenotypes, while endothelial dysfunction alters local mechanobiological signalling. Rather than facilitating complete repair, the extracellular matrix gradually stabilises the chronic adaptive state established during the initial injury response [28–42,51–69].
These structural adaptations have important consequences for microvascular physiology. Efficient oxygen extraction depends upon intimate spatial and functional coupling between endothelial cells, pericytes and the surrounding extracellular matrix. Progressive alterations in matrix composition, tissue stiffness and capillary geometry reduce the flexibility of the microcirculation, impair mechanotransduction and increase diffusion distances for oxygen and nutrients. Consequently, extracellular matrix remodelling further limits adaptive oxygen extraction while reinforcing endothelial–pericyte dysfunction [58–69].
The extracellular matrix also functions as a reservoir for numerous biologically active molecules. Cytokines, chemokines, growth factors and damage-associated molecular patterns may be retained within the matrix, where they continue to influence endothelial activation, immune-cell recruitment and tissue remodelling long after the initiating insult has subsided. This creates an additional mechanism through which persistent signalling may be maintained despite apparent resolution of the original injury [61–69].
Importantly, extracellular matrix remodelling should not be equated solely with fibrosis. Recovery failure is proposed to exist along a continuum of adaptive structural change ranging from subtle alterations in basement membrane organisation and tissue compliance to more advanced remodelling in longstanding disease. Much of this process is likely to remain biologically dynamic rather than permanently fixed, suggesting that restoration of physiological homeostasis may permit progressive normalisation of tissue architecture before irreversible fibrosis develops.
Within the present framework, the extracellular matrix therefore functions as the structural integrator of persistent adaptive signalling. By progressively embedding inflammatory, vascular and hypoxic adaptation into tissue architecture, ECM remodelling transforms a predominantly functional disturbance into a more stable biological state, reducing physiological flexibility and accelerating depletion of physiological reserve.
3.5 Failure of Tissue Clearance: Lymphatic, Glymphatic and Interstitial Dysfunction
Successful recovery depends not only upon restoration of oxygen delivery and tissue repair but also upon efficient removal of inflammatory mediators, cellular debris, metabolic by-products and excess interstitial fluid. These clearance processes are coordinated through the lymphatic, glymphatic and interstitial drainage systems, which function in close association with the microcirculation to restore tissue homeostasis following physiological stress. Failure of these integrated clearance pathways provides an additional mechanism through which persistent biological signalling may be sustained long after the initiating insult has resolved [70–77].
Within the central nervous system, the glymphatic system facilitates convective movement of cerebrospinal fluid through perivascular spaces, promoting clearance of metabolites, inflammatory mediators and cellular waste from the brain parenchyma. Meningeal lymphatic vessels subsequently transport these materials into the systemic lymphatic circulation. Efficient glymphatic function depends upon intact neurovascular coupling, normal vascular pulsatility, functional astrocytic water transport, preserved perivascular architecture and unobstructed lymphatic drainage. Disruption of any component of this integrated clearance pathway reduces the efficiency of waste removal and prolongs exposure of neural tissues to inflammatory and metabolic stress [70–77].
Comparable principles operate throughout the systemic circulation. Lymphatic vessels continuously regulate interstitial fluid balance, immune-cell trafficking and clearance of inflammatory mediators from peripheral tissues. Persistent endothelial activation, increased capillary permeability, extracellular matrix remodelling and chronic inflammatory signalling increase the burden placed upon the lymphatic system while simultaneously reducing its efficiency. Progressive interstitial congestion may therefore develop despite relatively preserved arterial perfusion, further compromising oxygen diffusion and adaptive tissue function [61–77].
Failure of tissue clearance should not be regarded as an isolated pathological process but as an integral component of the Recovery Failure framework. Persistent dysfunction of the endothelial–pericyte unit alters capillary permeability and interstitial fluid dynamics, while extracellular matrix remodelling changes tissue compliance and fluid movement.
These abnormalities reduce clearance of DAMPs, cytokines, reactive oxygen species and metabolic by-products, prolonging activation of TLR4, RAGE and downstream inflammatory pathways. Consequently, impaired clearance both results from and reinforces the Persistence Network, establishing an additional positive feedback mechanism that promotes chronic biological adaptation [16–46,61–77].
The concept of impaired clearance also provides a physiological explanation for the persistence of symptoms after apparent resolution of the initiating insult. Once infection or tissue injury has subsided, inefficient removal of inflammatory mediators and metabolic waste may continue to stimulate innate immune pathways, maintain endothelial activation and reinforce hypoxic adaptation despite the absence of ongoing tissue damage. Chronic illness therefore need not imply persistent infection or irreversible pathology, but may instead reflect failure of the integrated biological systems responsible for restoring tissue homeostasis.
Restoration of tissue clearance is therefore inseparable from restoration of microvascular function. Recovery requires re-establishment of coordinated endothelial–pericyte regulation, normalisation of extracellular matrix dynamics and recovery of lymphatic, glymphatic and interstitial drainage. Together these adaptive systems permit efficient removal of inflammatory mediators, optimise tissue oxygenation and progressively rebuild physiological reserve.
Accordingly, failure of tissue clearance represents the final component of the biological transition from persistent molecular signalling to impaired tissue physiology. Together with endothelial–pericyte dysfunction, impaired oxygen extraction and extracellular matrix remodelling, it establishes a self-reinforcing adaptive state that progressively erodes physiological reserve and prevents complete biological recovery.
Collectively, persistent molecular signalling, endothelial–pericyte dysfunction, impaired oxygen extraction, extracellular matrix remodelling and failure of tissue clearance transform an initially adaptive response into a chronically stabilised biological state. The functional consequence is progressive depletion of physiological reserve, which represents the central physiological expression of recovery failure.
4. Progressive Loss of Physiological Reserve: The Functional Expression of Recovery Failure
4.1 Physiological Reserve: The Foundation of Adaptive Biology
The biological processes described in the preceding sections converge upon a common physiological consequence: progressive depletion of physiological reserve. Persistent activation of the Persistence Network, endothelial–pericyte dysfunction, impaired adaptive oxygen extraction, extracellular matrix remodelling and failure of tissue clearance collectively reduce the capacity of biological systems to respond appropriately to increasing physiological demand. Within the Recovery Failure framework, this progressive loss of adaptive capacity represents the principal functional expression of chronic disease.
Health is characterised not simply by normal physiology at rest, but by the ability to adapt when physiological demand changes.
Physiological reserve is the integrated capacity of biological systems to increase functional performance above resting requirements in response to physiological stress while maintaining homeostasis and enabling complete recovery. It describes the capacity of an organism to increase biological performance above resting requirements in response to physical, cognitive, autonomic or environmental stress. Rather than representing the function of a single organ, reserve emerges from the coordinated interaction of cardiovascular, neurovascular, metabolic, inflammatory and structural regulatory systems.
It provides the functional link between molecular persistence and clinical disease. It represents the integrated biological capacity that enables organisms to tolerate stress, recover from injury and maintain homeostasis across multiple physiological systems. Consequently, depletion of reserve becomes the defining physiological hallmark of recovery failure.
Healthy physiology is therefore characterised not simply by normal resting function, but by the ability to adapt rapidly and efficiently when physiological demands change [81–91].
During normal activity, physiological reserve is continuously recruited. Exercise increases metabolic demand, orthostatic stress requires rapid cardiovascular adjustment, cognitive activity increases cerebral energy requirements, while infection activates inflammatory and reparative pathways. These challenges are accommodated through coordinated increases in cardiac output, capillary recruitment, oxygen extraction, substrate utilisation, autonomic regulation and inflammatory resolution. Once the stressor has resolved, these adaptive responses are progressively withdrawn and reserve is restored to its baseline state [81–91].
Recovery failure fundamentally alters this adaptive process. Persistent molecular signalling progressively impairs the biological systems responsible for maintaining reserve. Endothelial–pericyte dysfunction limits adaptive oxygen extraction, extracellular matrix remodelling reduces tissue flexibility, impaired clearance prolongs inflammatory activation and chronic hypoxic adaptation increases the energetic cost of maintaining homeostasis. Although basal physiological function may remain relatively preserved, progressively less reserve remains available to meet increasing physiological demand.
Importantly, physiological reserve should not be regarded as a static quantity but as a dynamic property that is continuously renewed through successful recovery. Reserve expands following restoration of adaptive function and contracts when recovery remains incomplete.
Chronic disease therefore reflects progressive depletion of adaptive reserve, rather than irreversible failure of individual organs. This distinction explains why patients often demonstrate relatively normal resting physiology while experiencing profound deterioration during even modest physical, cognitive or orthostatic stress.
4.2 Progressive Depletion of Adaptive Capacity
The Recovery Failure framework proposes that chronic illness develops through progressive depletion of adaptive capacity rather than progressive failure of individual organs. Persistent activation of the Persistence Network does not simply sustain inflammation or endothelial dysfunction; it progressively impairs the biological systems responsible for responding to physiological stress. Recovery failure should therefore be regarded as a disorder of adaptive physiology, in which the capacity to increase function becomes progressively limited despite relative preservation of basal physiological activity.
The earliest manifestation of reserve depletion occurs within the microcirculation. Persistent endothelial–pericyte dysfunction reduces the ability of capillary networks to adapt dynamically to changing metabolic demand. Although resting tissue perfusion may remain adequate, the capacity to recruit additional capillary exchange, optimise oxygen extraction and maintain metabolic homeostasis during physiological stress progressively declines. Consequently, adaptive oxygen extraction reserve becomes increasingly restricted, exposing tissues to intermittent functional hypoxia whenever physiological demand increases [47–60,89–91].
Reduced oxygen extraction initiates a cascade of secondary adaptive responses. Persistent HIF-dependent signalling promotes metabolic reprogramming towards glycolysis, while chronic activation of NF-κB and STAT3 maintains inflammatory and reparative programmes beyond their normal physiological duration. Extracellular matrix remodelling alters tissue mechanics and capillary architecture, and impaired lymphatic and glymphatic clearance prolongs exposure to inflammatory mediators and metabolic by-products. Collectively, these processes increase the energetic cost of maintaining homeostasis while simultaneously reducing the efficiency with which adaptive responses are generated [22–77].
An important feature of this model is that reserve depletion is cumulative. Every episode of infection, prolonged orthostatic stress, physical exertion, cognitive overload, surgery, trauma or significant inflammatory activation requires mobilisation of physiological reserve. When recovery is incomplete, adaptive systems do not fully return to baseline before the next physiological challenge occurs. Residual dysfunction therefore accumulates progressively, reducing the reserve available for subsequent stressors. Disease progression reflects repeated episodes of incomplete biological restoration rather than continuous tissue injury.
This cumulative model also explains the fluctuating nature of many chronic recovery-failure disorders. Physiological reserve is dynamic, not static. During periods of reduced physiological demand, the remaining reserve may be sufficient to maintain relatively normal function. However, as demand increases, the progressively diminished adaptive capacity is exceeded, producing disproportionate deterioration followed by delayed recovery. Clinical fluctuation therefore reflects continual movement around the threshold of available reserve rather than episodic recurrence of disease activity.
Importantly, depletion of adaptive capacity does not imply irreversible pathology. Early reserve loss is likely to reflect predominantly functional disturbances involving endothelial signalling, pericyte regulation, metabolic flexibility and inflammatory resolution. As persistent signalling continues, structural adaptation through extracellular matrix remodelling progressively stabilises the dysfunctional state, reducing physiological flexibility and slowing recovery. Thus, recovery failure evolves along a continuum from functional dysregulation to biologically embedded adaptation, rather than through abrupt transition to irreversible organ damage.
Progressive depletion of adaptive capacity provides the physiological explanation for why conventional resting investigations frequently underestimate disease severity. Basal physiological function may remain sufficient to satisfy resting metabolic requirements, while the capacity to increase performance during physical, cognitive or orthostatic stress becomes progressively constrained. The defining abnormality is therefore not impaired resting physiology, but loss of physiological responsiveness. Reserve depletion is cumulative because recovery is incomplete.
Accordingly, progressive depletion of adaptive capacity represents the critical transition between persistent molecular signalling and chronic multisystem disease. As reserve diminishes, biological resilience declines across multiple organ systems, progressively reducing the capacity to tolerate physiological stress and increasing the likelihood of prolonged recovery following even minor challenges.
4.3 Physiological Reserve as a Multisystem Property
Physiological reserve should not be regarded as a single physiological variable but as an emergent property arising from the coordinated interaction of multiple adaptive systems. The ability to tolerate physical, cognitive, autonomic and environmental stress depends upon simultaneous recruitment of cardiovascular, neurovascular, metabolic, inflammatory and structural mechanisms, each contributing a distinct component to overall biological resilience. Consequently, recovery failure does not result from dysfunction within a single organ but from progressive depletion of reserve across multiple interconnected physiological domains [81–91].
Each reserve domain fulfils a specialised adaptive role while remaining functionally integrated with the others. Haemodynamic reserve maintains adequate circulatory adaptation during changes in posture and exercise. Microvascular reserve regulates capillary recruitment and oxygen extraction. Neurovascular reserve preserves cerebral perfusion and neurovascular coupling. Metabolic reserve sustains ATP generation and substrate flexibility. Autonomic reserve coordinates rapid physiological responses, while inflammatory resolution reserve terminates adaptive inflammatory signalling and restores tissue homeostasis. Structural reserve, maintained through the extracellular matrix and supporting tissues, preserves the mechanical flexibility required for efficient physiological adaptation [47–91].
Importantly, these reserve systems do not function independently. Failure within one domain inevitably increases demands upon the others. For example, impaired microvascular oxygen extraction increases metabolic stress, amplifying inflammatory signalling through the Persistence Network.
Persistent inflammation further disrupts endothelial–pericyte communication, while extracellular matrix remodelling progressively reduces structural flexibility and impairs lymphatic clearance. This creates a self-reinforcing cycle in which depletion of one reserve domain accelerates deterioration across the entire adaptive system.
This integrated model provides a physiological explanation for the remarkable clinical heterogeneity observed across Long COVID, POTS, ME/CFS and related disorders. Individual patients may exhibit differing combinations of autonomic dysfunction, cognitive impairment, orthostatic intolerance, fatigue, sensory hypersensitivity or exercise limitation depending upon which reserve domains are preferentially affected. Despite these differing clinical phenotypes, the underlying biological process remains one of progressive loss of integrated adaptive capacity.
The concept of multisystem reserve also explains why disease severity frequently fluctuates. During periods of reduced physiological demand, the remaining reserve may be sufficient to maintain relative physiological stability. As physiological stress increases, however, progressively depleted reserve becomes exhausted, producing disproportionate deterioration across multiple physiological systems simultaneously. Clinical worsening therefore reflects temporary exceedance of available adaptive capacity rather than abrupt deterioration of individual organs.
Within the Recovery Failure framework, physiological reserve should therefore be regarded as the principal functional measure of health. Dynamic assessment of reserve across multiple biological domains is likely to provide a more meaningful evaluation of disease severity and recovery than isolated measurements obtained under resting conditions.
Table 2. Domains of Physiological Reserve
Reserve Domain | Primary Adaptive Function | Consequence of Reserve Depletion |
Haemodynamic Reserve | Maintains cardiac output, preload and circulatory adaptation during physiological stress | Orthostatic intolerance, reduced exercise capacity |
Microvascular Reserve | Regulates capillary recruitment and adaptive oxygen extraction | Functional tissue hypoxia, impaired oxygen utilisation |
Neurovascular Reserve | Maintains cerebral perfusion and neurovascular coupling | Cognitive dysfunction, impaired concentration, sensory hypersensitivity |
Metabolic Reserve | Preserves ATP production and metabolic flexibility | Fatigue, impaired recovery, reduced exercise tolerance |
Autonomic Reserve | Coordinates cardiovascular, respiratory and visceral adaptation | Dysautonomia, impaired physiological responsiveness |
Inflammatory Resolution Reserve | Terminates inflammatory responses and restores tissue homeostasis | Persistent inflammatory activation and delayed recovery |
Structural Reserve | Maintains extracellular matrix flexibility, tissue compliance and mechanical integrity | Progressive tissue stiffening, impaired adaptive flexibility |
Figure 2. Adaptive microvascular reserve as the central integrator of physiological reserve.
Within the Recovery Failure framework, the endothelial–pericyte unit regulates adaptive microvascular reserve by coordinating capillary recruitment, oxygen extraction and microvascular adaptation. Progressive impairment of adaptive microvascular reserve reduces haemodynamic, neurovascular, metabolic, autonomic, inflammatory resolution and structural reserve, leading to multisystem physiological reserve loss and ultimately recovery failure.
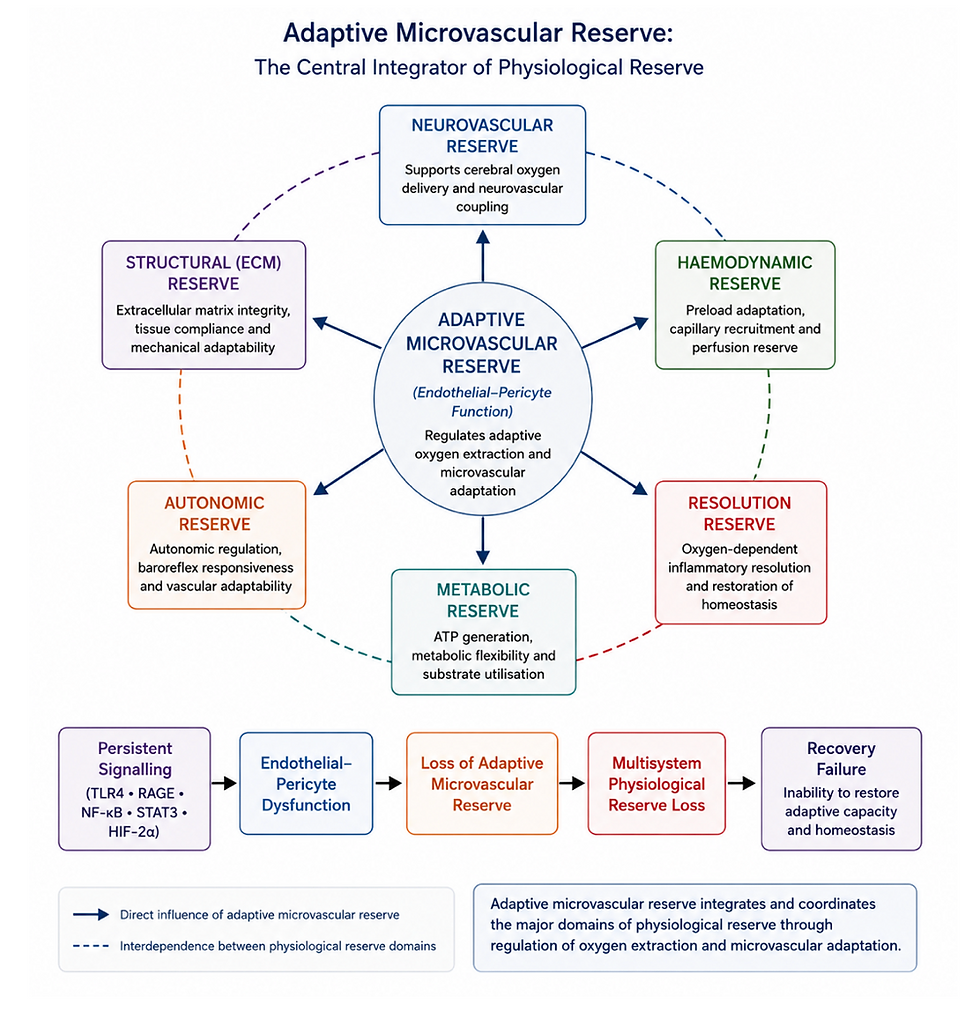
4.4 Clinical Expression of Progressive Reserve Loss
Progressive depletion of physiological reserve provides a unifying physiological explanation for the diverse clinical manifestations observed across Long COVID, POTS, ME/CFS and related recovery-failure disorders. Rather than reflecting isolated dysfunction of individual organs, symptoms emerge when the integrated adaptive systems responsible for maintaining physiological homeostasis are unable to meet increasing physiological demand. The clinical phenotype therefore reflects failure of adaptive capacity rather than failure of basal physiological function [1–15,81–91].
In healthy individuals, physical activity, orthostatic challenge, cognitive effort, thermal stress, infection and emotional stress all recruit physiological reserve through coordinated activation of cardiovascular, neurovascular, autonomic, metabolic and inflammatory systems. Endothelial–pericyte regulation increases capillary recruitment and oxygen extraction, autonomic responses redistribute blood flow, metabolic pathways increase ATP production and inflammatory pathways facilitate tissue repair. These adaptive responses occur rapidly and resolve efficiently once the physiological challenge has passed [47–60,81–91].
As physiological reserve progressively declines, increasingly modest stressors become sufficient to exceed the remaining adaptive capacity. Physical exertion, prolonged standing, cognitive activity, sensory stimulation, infection or emotional stress all increase physiological demand through different mechanisms, yet each ultimately requires mobilisation of the same integrated reserve systems. When reserve is inadequate, tissue oxygen extraction, metabolic flexibility and autonomic regulation can no longer be augmented sufficiently to maintain homeostasis, resulting in disproportionate symptom exacerbation followed by delayed biological recovery.
This framework explains the characteristic variability observed in many patients. During periods of low physiological demand, residual reserve may be sufficient to maintain relatively normal function. As demand increases, however, progressively depleted reserve becomes exhausted, producing multisystem deterioration despite relatively unchanged structural pathology. Symptom fluctuation therefore reflects continual variation in the relationship between physiological demand and available reserve, rather than intermittent activation of disease.
Clinical heterogeneity similarly becomes understandable within this model. Individuals with predominant depletion of haemodynamic reserve may present with orthostatic intolerance, exercise limitation and impaired cardiovascular adaptation. Greater loss of neurovascular reserve may manifest as cognitive dysfunction, headache, sensory hypersensitivity or impaired cerebral autoregulation.
Predominant depletion of metabolic reserve may present with fatigue and impaired recovery, whereas failure of inflammatory resolution reserve may prolong inflammatory symptoms and delay tissue repair. Structural reserve loss contributes to reduced mechanical flexibility and impaired tissue adaptation. Although these clinical phenotypes differ considerably, each reflects preferential expression of the same underlying process—progressive depletion of integrated physiological reserve.
Reserve depletion also provides a physiological explanation for the progressive narrowing of activity tolerance commonly reported by patients. Activities that were previously performed without difficulty increasingly exceed the available adaptive capacity, while recovery following relatively minor physiological stress becomes progressively prolonged. Disease progression therefore reflects progressive contraction of the adaptive range rather than abrupt deterioration of individual organ systems.
Within the Recovery Failure framework, symptom severity is determined less by the magnitude of persistent molecular signalling than by the degree to which physiological reserve has been depleted. Restoration of reserve therefore becomes the principal functional objective of recovery, providing a common endpoint through which diverse therapeutic approaches may ultimately be evaluated.
Table 3. Clinical Manifestations of Reserve Depletion
Predominant Reserve Domain Affected | Typical Clinical Expression | Underlying Physiological Disturbance |
Haemodynamic Reserve | Orthostatic intolerance, reduced exercise capacity, exertional dizziness | Impaired preload adaptation and circulatory reserve |
Microvascular Reserve | Exertional intolerance, tissue hypoxia, delayed recovery | Reduced adaptive oxygen extraction |
Neurovascular Reserve | Cognitive dysfunction, headache, sensory hypersensitivity, "brain fog" | Impaired neurovascular coupling and cerebral oxygen delivery |
Metabolic Reserve | Fatigue, reduced endurance, impaired recovery following physiological stress | Reduced metabolic flexibility and ATP generation |
Autonomic Reserve | Heart rate variability abnormalities, dysautonomia, impaired stress responses | Reduced autonomic adaptability |
Inflammatory Resolution Reserve | Persistent inflammatory symptoms, prolonged recovery after infection or injury | Failure of inflammatory resolution |
Structural Reserve | Reduced tissue flexibility, chronic musculoskeletal symptoms, progressive functional limitation | Extracellular matrix remodelling and impaired tissue mechanics |
Symptoms do not measure disease activity directly; they reflect the point at which physiological demand exceeds the remaining adaptive reserve.
4.5 Physiological Reserve as the Functional Endpoint of Recovery Failure
The preceding sections describe how persistent molecular signalling is translated into progressive impairment of tissue physiology through dysfunction of the endothelial–pericyte unit, impaired adaptive oxygen extraction, extracellular matrix remodelling and failure of tissue clearance.
Collectively, these interacting processes converge upon a single physiological consequence: progressive depletion of physiological reserve. Within the Recovery Failure framework, loss of reserve represents the principal functional endpoint through which persistent biological adaptation is expressed clinically.
This perspective differs fundamentally from traditional models that focus primarily upon isolated pathological mechanisms or individual organ dysfunction. Rather than proposing separate explanations for fatigue, autonomic dysfunction, cognitive impairment, exercise intolerance or delayed recovery, the present framework suggests that these diverse clinical manifestations arise because multiple adaptive systems progressively lose the capacity to respond appropriately to increasing physiological demand. Symptoms therefore represent differing expressions of a common physiological process rather than independent disease mechanisms [1–15,81–91].
Importantly, physiological reserve is proposed to decline progressively rather than catastrophically. During the early stages of disease, remaining reserve may compensate sufficiently to maintain near-normal function under resting conditions. As reserve becomes progressively depleted, however, the margin between physiological demand and adaptive capacity narrows. Everyday activities that were previously well tolerated increasingly exceed the available reserve, resulting in disproportionate symptom exacerbation and progressively prolonged recovery. Recovery failure therefore represents a continuum of diminishing adaptive flexibility rather than a binary transition between health and disease.
This model also provides a biological explanation for the convergence of apparently distinct chronic disorders. Long COVID, POTS, ME/CFS and related syndromes differ in their initiating events and dominant clinical manifestations, yet each appears capable of converging upon the same final common pathway characterised by persistent adaptive signalling, endothelial–pericyte dysfunction, impaired oxygen extraction and progressive depletion of physiological reserve. The predominant clinical phenotype reflects the relative contribution of different reserve domains rather than fundamentally different biological mechanisms.
Recognition of physiological reserve as the principal functional endpoint of recovery failure has important implications for both research and clinical practice. Conventional investigations predominantly assess resting physiology, whereas the defining abnormality proposed in this framework is impairment of adaptive physiology.
Dynamic assessment of haemodynamic adaptation, cerebral perfusion, microvascular function, oxygen extraction, autonomic regulation and recovery kinetics is therefore likely to provide a more meaningful evaluation of disease severity than isolated resting measurements [89–113].
Equally important, this framework shifts the therapeutic objective from suppression of individual molecular pathways to restoration of adaptive capacity. Successful recovery requires progressive rebuilding of the biological systems responsible for physiological flexibility, allowing tissues once again to increase function appropriately in response to physical, cognitive, autonomic and environmental stress. Recovery should therefore be assessed not solely by reduction in symptoms, but by restoration of physiological reserve and resilience.
Accordingly, physiological reserve provides the central organising principle of the Recovery Failure framework. The Persistence Network explains why adaptive signalling persists; the endothelial–pericyte unit explains how persistent signalling is translated into impaired tissue physiology; and progressive depletion of physiological reserve explains why patients lose resilience and develop chronic multisystem illness. Together these concepts provide a unified systems biology model linking molecular mechanisms with clinical disease expression.
The recognition that recovery failure reflects progressive loss of adaptive capacity rather than irreversible organ dysfunction also provides the conceptual foundation for the next section. If chronic illness represents a dynamic state of impaired adaptation, then disease progression should likewise be viewed as a dynamic biological process rather than a fixed clinical diagnosis.
5. Disease Evolution: From Adaptive Recovery to Recovery Failure
5.1 Recovery Failure as a Progressive Biological Process
Recovery following physiological injury is not a binary event but a dynamic biological process in which multiple adaptive systems progressively restore tissue homeostasis, structural integrity and physiological reserve. Following infection, trauma or other physiological stressors, inflammatory, vascular, metabolic and neuroendocrine pathways are activated to preserve tissue viability while initiating repair. Under normal circumstances these adaptive responses are self-limiting, resolving as tissue homeostasis is restored and physiological reserve returns to baseline [16–20,38–42,92–97].
Within the Recovery Failure framework, chronic illness develops when this restorative process remains incomplete. Persistent activation of the Persistence Network prevents full resolution of inflammatory, vascular and hypoxic adaptation, progressively impairing endothelial–pericyte function, limiting adaptive oxygen extraction and reducing restoration of physiological reserve. Recovery failure should therefore be viewed as a disorder of incomplete biological resolution, rather than persistent tissue injury alone.
This distinction reframes chronic disease. The Persistence Network is not inherently pathological; rather, it comprises adaptive programmes that have remained chronically engaged beyond their normal physiological role. Biological systems originally designed to preserve tissue survival progressively become maladaptive, reducing physiological flexibility while increasing the energetic cost of maintaining homeostasis.
This concept also explains why recovery frequently occurs along a continuum rather than through abrupt transitions. Patients may improve or deteriorate gradually as the balance between adaptive restoration and persistent signalling changes over time. Disease progression therefore reflects progressive movement along a recovery trajectory, not progression through rigid pathological stages.
5.2 Functional Adaptation Precedes Structural Adaptation
One of the central concepts emerging from this framework is that chronic recovery failure evolves through two broad phases of biological adaptation.
During the early phase, abnormalities are predominantly functional. Persistent inflammatory signalling alters endothelial regulation, pericyte behaviour, autonomic control, oxygen extraction and metabolic flexibility without necessarily producing major structural tissue injury. At this stage, adaptive physiology remains impaired but retains considerable potential for recovery because tissue architecture has not yet undergone substantial remodelling.
As persistent signalling continues, however, biological adaptation becomes progressively stabilised through extracellular matrix remodelling, altered tissue mechanics, changes in capillary architecture and impaired clearance of inflammatory mediators. These structural adaptations reduce physiological flexibility and increasingly reinforce the persistent adaptive state, making spontaneous recovery progressively less likely [61–69].
Importantly, this transition should not be interpreted as inevitable progression to irreversible fibrosis. Rather, recovery failure exists along a continuum ranging from reversible functional dysregulation to progressively stabilised biological adaptation. The position of an individual patient along this continuum is likely to influence both clinical severity and responsiveness to therapeutic intervention.
5.3 Progressive Contraction of Adaptive Capacity
As recovery becomes progressively incomplete, physiological reserve declines across multiple interacting systems. Consequently, the range of physiological challenges that can be accommodated without symptom exacerbation progressively contracts.
Initially, only substantial physiological stress exceeds the available reserve. Over time, however, increasingly modest physical activity, cognitive effort, orthostatic stress, infection, thermal exposure or emotional stress become sufficient to provoke deterioration because the remaining adaptive capacity has become progressively depleted.
This phenomenon may be viewed as progressive contraction of the adaptive range. Health is characterised by broad physiological flexibility, allowing efficient adaptation across a wide range of environmental conditions. Recovery failure progressively narrows this adaptive range until routine daily activities approach or exceed the available physiological reserve.
This concept provides a physiological explanation for the characteristic reduction in functional capacity reported by many patients and explains why symptom severity often fluctuates despite relatively stable structural pathology.
5.4 Biological Drivers of Disease Progression
Disease progression is unlikely to result from persistent signalling alone. Instead, repeated physiological challenges continually interact with an already compromised adaptive system, reinforcing the Persistence Network while further reducing physiological reserve.
Potential drivers include:
recurrent infection
prolonged orthostatic stress
repeated physical overexertion
sustained cognitive demand
surgery and tissue trauma
connective tissue instability
vascular compression syndromes
metabolic stress
environmental toxins
chronic neuroendocrine stress responses
Although these factors differ substantially in origin, each increases physiological demand while requiring mobilisation of adaptive reserve. When recovery remains incomplete, successive physiological challenges progressively reinforce the persistent adaptive state, producing cumulative biological deterioration.
5.5 The Recovery Trajectory
Rather than viewing chronic illness as a fixed disease state, the Recovery Failure framework proposes that patients exist along a dynamic biological recovery trajectory.
At one end of this trajectory, adaptive signalling resolves efficiently, endothelial–pericyte regulation is restored, oxygen extraction normalises, extracellular matrix remodelling resolves and physiological reserve returns to baseline.
At the opposite end, persistent signalling, impaired oxygen extraction, extracellular matrix remodelling and clearance failure become increasingly self-reinforcing, producing progressive depletion of physiological reserve and chronic multisystem dysfunction.
Importantly, movement along this trajectory is not necessarily unidirectional. Because adaptive biology remains inherently dynamic, restoration of endothelial–pericyte function, improvement in oxygen extraction, recovery of tissue clearance and rebuilding of physiological reserve may permit gradual movement back towards health. The extent of recovery is therefore likely to depend upon the degree of structural adaptation that has occurred and the residual capacity of adaptive systems to regain physiological flexibility.
Accordingly, recovery should not be viewed simply as symptom reduction but as progressive restoration of adaptive biology. This concept provides the biological bridge to the next section, which considers the fundamental principles governing recovery and restoration of physiological reserve.
Figure 3. The Recovery Trajectory.
Recovery following physiological injury normally progresses through coordinated adaptive responses that restore homeostasis and physiological reserve. Within the Recovery Failure framework, persistent signalling redirects this trajectory towards endothelial–pericyte dysfunction, impaired oxygen extraction, extracellular matrix remodelling, clearance failure and progressive loss of physiological reserve. Disease progression is therefore viewed as a dynamic continuum of incomplete biological resolution rather than irreversible organ failure. Importantly, recovery failure is not a fixed destination but a potentially modifiable biological trajectory, in which restoration of adaptive physiology may stabilise, slow or reverse progression.

6. Recovery Biology: Restoring Adaptive Capacity
6.1 Recovery is an Active Biological Process
Recovery is often viewed as the passive consequence of resolution of disease. However, restoration of health is itself an active biological process requiring coordinated regulation of inflammatory signalling, vascular adaptation, tissue repair and metabolic homeostasis. Recovery therefore depends not simply upon removal of the initiating insult but upon re-establishing the adaptive systems that restore physiological flexibility and rebuild physiological reserve.
Within the Recovery Failure framework, persistent illness develops because these restorative mechanisms remain incomplete. Inflammatory signalling, endothelial activation, hypoxic adaptation and extracellular matrix remodelling continue beyond their normal physiological purpose, maintaining tissues in a chronic adaptive state designed for survival rather than restoration. Recovery therefore requires active re-establishment of physiological homeostasis rather than passive disappearance of symptoms [16–20,22–46,92–97].
This distinction fundamentally changes how chronic disease is interpreted. Recovery is not simply the absence of inflammation or the correction of isolated biochemical abnormalities. Rather, it represents progressive restoration of the integrated biological systems responsible for adaptive regulation across the entire organism.
6.2 Resolution Rather Than Suppression
An important implication of this framework is the distinction between resolution and suppression.
Inflammatory pathways such as TLR4, NF-κB, STAT3 and HIF signalling are essential components of normal host defence and tissue repair. Their complete suppression would impair physiological adaptation and increase susceptibility to infection or injury. The biological objective is therefore not elimination of these pathways but restoration of their normal physiological regulation.
Resolution involves coordinated termination of persistent signalling once tissue repair has been completed. Endothelial–pericyte communication must recover, adaptive oxygen extraction must normalise, extracellular matrix remodelling must return towards physiological balance, and lymphatic, glymphatic and interstitial clearance systems must once again efficiently remove inflammatory mediators and metabolic by-products. These coordinated processes progressively restore physiological flexibility and rebuild reserve [92–97].
This concept explains why symptomatic improvement does not necessarily indicate biological recovery. A reduction in symptoms may occur while adaptive physiology remains substantially impaired. Conversely, restoration of adaptive reserve may precede complete symptom resolution as physiological resilience gradually returns.
6.3 Rebuilding the Adaptive Systems
Recovery requires coordinated restoration of multiple interacting biological systems.
· The Persistence Network must progressively return to physiological regulation, allowing inflammatory signalling to terminate appropriately while preserving normal host defence.
The endothelial–pericyte unit must recover its ability to regulate capillary recruitment, endothelial permeability and adaptive oxygen extraction.
Extracellular matrix remodelling must gradually restore tissue flexibility and normal mechanobiology.
Lymphatic, glymphatic and interstitial clearance systems must regain efficient removal of inflammatory mediators, damaged proteins and metabolic waste products.
Finally, physiological reserve must progressively expand across haemodynamic, microvascular, neurovascular, autonomic, metabolic, inflammatory resolution and structural domains.
Importantly, these processes occur simultaneously rather than sequentially. Improvement in one adaptive system facilitates recovery of the others, illustrating that recovery itself is fundamentally a systems-level biological phenomenon rather than the repair of isolated molecular abnormalities.
6.4 Restoration of Physiological Flexibility
Perhaps the defining characteristic of successful recovery is restoration of physiological flexibility.
Healthy organisms continuously adjust cardiovascular performance, capillary recruitment, oxygen extraction, autonomic activity, metabolism and inflammatory responses according to changing physiological demand. These adaptive responses occur rapidly, efficiently and with minimal energetic cost.
Recovery failure progressively narrows this adaptive range. Patients frequently describe an increasingly restricted capacity to tolerate physical activity, cognitive effort, orthostatic stress, environmental change or infection before deterioration occurs. Successful recovery therefore requires gradual expansion of this adaptive range through restoration of physiological reserve rather than simple reduction of resting symptoms.
Within this framework, restoration of flexibility becomes the functional expression of biological recovery. Increasing tolerance of physiological stress reflects expansion of adaptive reserve and progressive recovery of the integrated biological systems responsible for maintaining homeostasis.
6.5 Measuring Recovery
If recovery is fundamentally the restoration of adaptive biology, then the methods used to evaluate recovery should also change.
Traditional investigations largely assess static physiological variables measured under resting conditions. While these measurements remain clinically important, they provide limited information regarding the adaptive performance of biological systems.
The Recovery Failure framework predicts that future biomarkers of recovery will increasingly focus upon dynamic physiological performance, including:
restoration of endothelial–pericyte function;
improvement in adaptive oxygen extraction;
recovery of haemodynamic and neurovascular reserve;
restoration of autonomic adaptability;
normalisation of inflammatory resolution;
expansion of physiological reserve during physical, cognitive and orthostatic challenge.
Dynamic assessment of recovery kinetics, rather than isolated resting measurements, may therefore provide more meaningful indicators of biological restoration and therapeutic efficacy [81–97].
Accordingly, recovery biology represents a natural extension of systems medicine. Rather than focusing solely upon disease initiation or suppression of persistent signalling, it emphasises restoration of adaptive physiology as the central objective of clinical management and future therapeutic development.
7. Conclusion
Recovery following infection, injury and physiological stress has traditionally been viewed as the passive consequence of disease resolution. This review proposes an alternative perspective. Rather than occurring automatically once the initiating insult has subsided, recovery is presented as an active, highly coordinated biological process requiring restoration of inflammatory regulation, microvascular adaptation, oxygen extraction, extracellular matrix homeostasis, tissue clearance and physiological reserve.
Chronic illness is therefore interpreted not simply as persistence of disease, but as failure of the biological systems responsible for restoring physiological homeostasis.
Central to this framework is the concept of the Persistence Network, an integrated adaptive signalling system comprising TLR4, RAGE, NF-κB, STAT3, CCL2 and hypoxia-inducible pathways. While each pathway fulfils an essential physiological role during acute injury and host defence, persistent activation transforms these normally protective responses into a self-reinforcing adaptive state capable of sustaining inflammatory, vascular, metabolic and hypoxic signalling long after the initiating insult has resolved.
The biological importance of the Persistence Network lies not in the individual pathways themselves, but in the emergent behaviour generated through their continuous interaction.
Persistent signalling is proposed to converge upon the endothelial–pericyte unit, the principal adaptive regulator of the microcirculation. Progressive disruption of endothelial–pericyte communication impairs adaptive oxygen extraction, promotes extracellular matrix remodelling, alters tissue clearance and progressively reduces the efficiency with which tissues respond to increasing physiological demand. These interacting processes provide a biological explanation for the widespread yet often subtle physiological abnormalities observed across Long COVID, POTS, ME/CFS and related disorders.
The central physiological consequence of these changes is progressive depletion of physiological reserve. Rather than representing isolated failure of individual organs, chronic disease is proposed to reflect progressive loss of adaptive capacity across haemodynamic, microvascular, neurovascular, metabolic, autonomic, inflammatory and structural domains. Symptoms emerge when physiological demand exceeds the remaining reserve, explaining the characteristic exercise intolerance, orthostatic dysfunction, cognitive impairment and delayed recovery that unify these otherwise diverse clinical conditions.
This framework also redefines the biological objective of recovery. Resolution of persistent illness requires more than suppression of inflammatory pathways or correction of isolated physiological abnormalities. Successful recovery is proposed to depend upon restoration of adaptive biology itself, including normal regulation of the Persistence Network, recovery of endothelial–pericyte function, restoration of adaptive oxygen extraction, normalisation of extracellular matrix dynamics and tissue clearance, and progressive rebuilding of physiological reserve. Recovery is therefore best understood as restoration of physiological flexibility, enabling biological systems once again to adapt efficiently to changing physiological demand.
Although many aspects of this framework remain hypothetical and require prospective validation, it provides a coherent systems biology model that integrates molecular signalling, microvascular physiology, tissue remodelling and whole-organism adaptation within a single conceptual structure. By shifting attention from individual disease mechanisms towards the biology of recovery itself, the Recovery Failure framework offers a unifying explanation for the shared pathophysiology of Long COVID, POTS, ME/CFS and related disorders while providing a foundation for future mechanistic research and development of therapies directed towards restoration of adaptive physiology rather than suppression of persistent disease alone.
References
Sheldon RS, Grubb BP, Olshansky B, Shen WK, Calkins H, Brignole M, et al. 2015 Heart Rhythm Society expert consensus statement on postural tachycardia syndrome, inappropriate sinus tachycardia and vasovagal syncope. Heart Rhythm. 2015;12(6):e41–e63.
Raj SR, Guzman JC, Harvey P, Richer L, Schondorf R, Seifer C, et al. Canadian Cardiovascular Society position statement on postural orthostatic tachycardia syndrome and related disorders of chronic orthostatic intolerance. Can J Cardiol. 2020;36(3):357–372.
Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21:133–146.
Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. 2021;27(4):601–615.
Peluso MJ, Deeks SG. Mechanisms of long COVID and the path toward therapeutics. Cell. 2024;187:3401–3424.
Astin R, Banerjee A, Baker MR, Dani M, Ford E, Hull JH, et al. Long COVID: mechanisms, risk factors and recovery. Exp Physiol. 2023;108(1):12–27.
Komaroff AL, Lipkin WI. Insights from myalgic encephalomyelitis/chronic fatigue syndrome may help unravel the pathogenesis of postacute COVID-19 syndrome. Trends Mol Med. 2021;27(9):895–906.
Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19: an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. 2021;12:698169.
National Institute for Health and Care Excellence. Myalgic encephalomyelitis/chronic fatigue syndrome: diagnosis and management. NICE guideline NG206. London: NICE; 2021.
National Academies of Sciences, Engineering, and Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington, DC: National Academies Press; 2015.
Hickie I, Davenport T, Wakefield D, Vollmer-Conna U, Cameron B, Vernon SD, et al. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ. 2006;333:575.
Carruthers BM, Jain AK, De Meirleir KL, Peterson DL, Klimas NG, Lerner AM, et al. Myalgic encephalomyelitis/chronic fatigue syndrome: clinical working case definition, diagnostic and treatment protocols. J Chronic Fatigue Syndr. 2003;11(1):7–115.
Carruthers BM, van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, et al. Myalgic encephalomyelitis: international consensus criteria. J Intern Med. 2011;270(4):327–338.
Lim EJ, Ahn YC, Jang ES, Lee SW, Lee SH, Son CG. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis. J Transl Med. 2020;18:100.
Jason LA, Richman JA, Rademaker AW, Jordan KM, Plioplys AV, Taylor RR, et al. A community-based study of chronic fatigue syndrome. Arch Intern Med. 1999;159:2129–2137.
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity. Nat Immunol. 2010;11:373–384.
Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162.
Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. 2017;14(1):43–64.
Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021;31:818–820.
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388.
Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE: signalling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. 2011;1243:88–102.
Pickering RJ, Tikellis C, Rosado CJ, Tsorotes D, Dimitropoulos A, Smith M, et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J Clin Invest. 2019;129(1):406–421.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–913.
Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, et al. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18(10):1342–1352.
Leclerc E, Sturchler E, Vetter SW. The S100B/RAGE axis in Alzheimer’s disease. Cardiovasc Psychiatry Neurol. 2010;2010:539581.
Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023.
Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–234.
Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–324.
Pasparakis M. Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–788.
Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. NF-κB in monocytes and macrophages — an inflammatory master regulator in multitalented immune cells. Front Immunol. 2023;14:1134661.
Capece D, Verzella D, Di Francesco B, Alesse E, Franzoso G, Zazzeroni F. NF-κB: blending metabolism, immunity and inflammation. Trends Immunol. 2022;43:757–775.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to inflammatory and fibrotic diseases and prospects for its targeting for treatment. Int J Mol Sci. 2018;19(8):2299.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448–457.
Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106:365–374.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408.
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402.
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22.
Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer. 2016;2(12):758–770.
Bandarra D, Biddlestone J, Mudie S, Müller HAJ, Rocha S. HIF-1α restricts NF-κB-dependent gene expression to control innate immunity signals. Dis Model Mech. 2015;8:169–181.
Singh S, Anshita D, Ravichandiran V. MCP-1: function, regulation and involvement in disease. Int Immunopharmacol. 2021;101:107598.
Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilisation from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117(4):902–909.
Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–889.
Rose CE Jr, Sung SSJ, Fu SM. Significant involvement of CCL2/MCP-1 in inflammatory disorders. Microcirculation. 2003;10(3–4):273–288.
Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376.
Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243.
Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke and Alzheimer disease. J Appl Physiol. 2006;100(1):328–335.
Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–1405.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signalling pathways. Nat Neurosci. 2016;19(6):771–783.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738.
Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057–1069.
Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2012;2:a006429.
Pries AR, Secomb TW. Making microvascular networks work: angiogenesis, remodelling and adaptation. Microcirculation. 2014;21:315–331.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Augustin HG, Koh GY. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science. 2017;357:eaal2379.
Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200.
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27.
Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–812.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801.
Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodelling in development and disease. Cold Spring Harb Perspect Biol. 2011;3:a005058.
Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10:712–723.
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210.
Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309–326.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes. Sci Transl Med. 2012;4:147ra111.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–999.
Mestre H, Mori Y, Nedergaard M. The brain's glymphatic system: current controversies. Trends Neurosci. 2020;43:458–466.
Nedergaard M. Garbage truck of the brain. Science. 2013;340:1529–1530.
Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50:3–20.
Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21st century: novel functional roles in homeostasis and disease. Cell. 2020;182:270–296.
Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353:766–771.
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–477.
Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation. Nat Rev Immunol. 2019;19:477–489.
Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an ^11C-(R)-PK11195 PET study. J Nucl Med. 2014;55:945–950.
Cannon WB. The Wisdom of the Body. New York: W.W. Norton; 1932.
Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Fisher S, Reason J, editors. Handbook of Life Stress, Cognition and Health. New York: John Wiley & Sons; 1988. p. 629–649.
McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci. 2006;8:367–381.
McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43:2–15.
Goldstein DS. Concepts of scientific integrative medicine applied to the physiology and pathophysiology of catecholamine systems. Compr Physiol. 2020;10:1–75.
Lipsitz LA. Physiological complexity, aging and the path to frailty. Sci Aging Knowledge Environ. 2004;16:pe16.
Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664.
Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837.
Guyton AC, Hall JE. Textbook of Medical Physiology. 14th ed. Philadelphia: Elsevier; 2020.
Joyner MJ, Casey DP. Regulation of increased blood flow during exercise: a hierarchy of competing physiological needs. Physiol Rev. 2015;95:549–601.
Magder S. Volume and its relationship to cardiac output and venous return. Crit Care. 2016;20:271.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101.
Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128:2657–2669.
Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–567.
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882.
Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172.
Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13:59–66.
van Campen CLMC, Rowe PC, Visser FC. Cerebral blood flow is reduced in severe myalgic encephalomyelitis/chronic fatigue syndrome during mild orthostatic stress testing. Clin Neurophysiol Pract. 2020;5:50–58.
Barnden LR, Crouch B, Kwiatek R, Burnet R, Del Fante P, Guo J, et al. A brain MRI study of chronic fatigue syndrome: evidence of brainstem dysfunction and altered myelination. NMR Biomed. 2015;28:895–902.
Fluge Ø, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 2016;1:e89376.
Wirth K, Scheibenbogen C. A unifying hypothesis of the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome. Front Pediatr. 2020;8:124.
Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, et al. Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci U S A. 2016;113:E5472–E5480.
Tomas C, Brown A, Strassheim V, Elson JL, Newton J, Manning P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS One. 2017;12:e0186802.
Cotler J, Holtzman C, Dudun C, Jason LA. A brief questionnaire to assess post-exertional malaise. Diagnostics (Basel). 2018;8:66.
Goldstein DS. The extended autonomic system, dyshomeostasis, and COVID-19. Clin Auton Res. 2020;30:299–315.
Seals DR, Esler MD. Human ageing and the sympathoadrenal system. J Physiol. 2000;528:407–417.
Benarroch EE. The central autonomic network: functional organisation, dysfunction and perspective. Mayo Clin Proc. 1993;68:988–1001.
Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346.
Dampney RAL. Central neural control of the cardiovascular system: current perspectives. Adv Physiol Educ. 2016;40:283–296.
Saper CB. The central autonomic nervous system: conscious visceral perception and autonomic pattern generation. Annu Rev Neurosci. 2002;25:433–469.
Vernino S, Bourne KM, Stiles LE, Grubb BP, Fedorowski A, Stewart JM, et al. Postural orthostatic tachycardia syndrome: state of the science and clinical care. Auton Neurosci. 2021;235:102828.
Stewart JM. Common syndromes of orthostatic intolerance. Pediatrics. 2013;131:968–980.
Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and postural tachycardia syndrome. Clin Auton Res. 2011;21:69–72.
Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular and systemic manifestations. ISRN Dermatol. 2012;2012:751768.
Henderson FC Sr, Austin C, Benzel E, Bolognese P, Ellenbogen R, Francomano CA, et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:195–211.
Beggs CB. Venous hemodynamics in neurological disorders: an analytical review with hydrodynamic analysis. BMC Med. 2013;11:142.
Doepp F, Schreiber SJ, von Münster T, Rademacher J, Klingebiel R, Valdueza JM. How does the blood leave the brain? A systematic ultrasound analysis of cerebral venous drainage patterns. Neuroradiology. 2004;46:565–570.
Zhao H, Alam A, Chen Q, Eusman MA, Pal A, Eguchi S, et al. The role of microglia in the pathobiology of neuropathic pain development: what do we know? Br J Anaesth. 2017;118:504–516.
Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signalling contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2014;15:712–725.
Lees JG, Makker PGS, Tonkin RS, Abdulla M, Park SB, Goldstein D, et al. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur J Cancer. 2017;73:22–29.
Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, et al. Incidence, prevalence and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain. 2014;155:2461–2470.
Chovatiya R, Medzhitov R. Stress, inflammation, and defence of homeostasis. Mol Cell. 2014;54:281–288.
Medzhitov R. Disease tolerance as a defence strategy. Science. 2012;335:936–941.
Comments